According to the U.S. FDA’s traditional “Weight of Evidence” approach, in order to demonstrate bioequivalence (BE), the developer must be able to demonstrate that the formulation and device are similar, in vitro tests are equivalent, systemic pharmacokinetic (PK) exposure is equivalent and pharmacodynamics (PD) or comparative clinical endpoint (CCEP) efficacy are equivalent.
Since 2024, the need for a CCEP study has been no longer mandatory in lieu of additional in vitro and PK studies. This change appears to reduce the burden on the developer, but in practice has led to other challenges which have arisen following the implementation of the additional tests.
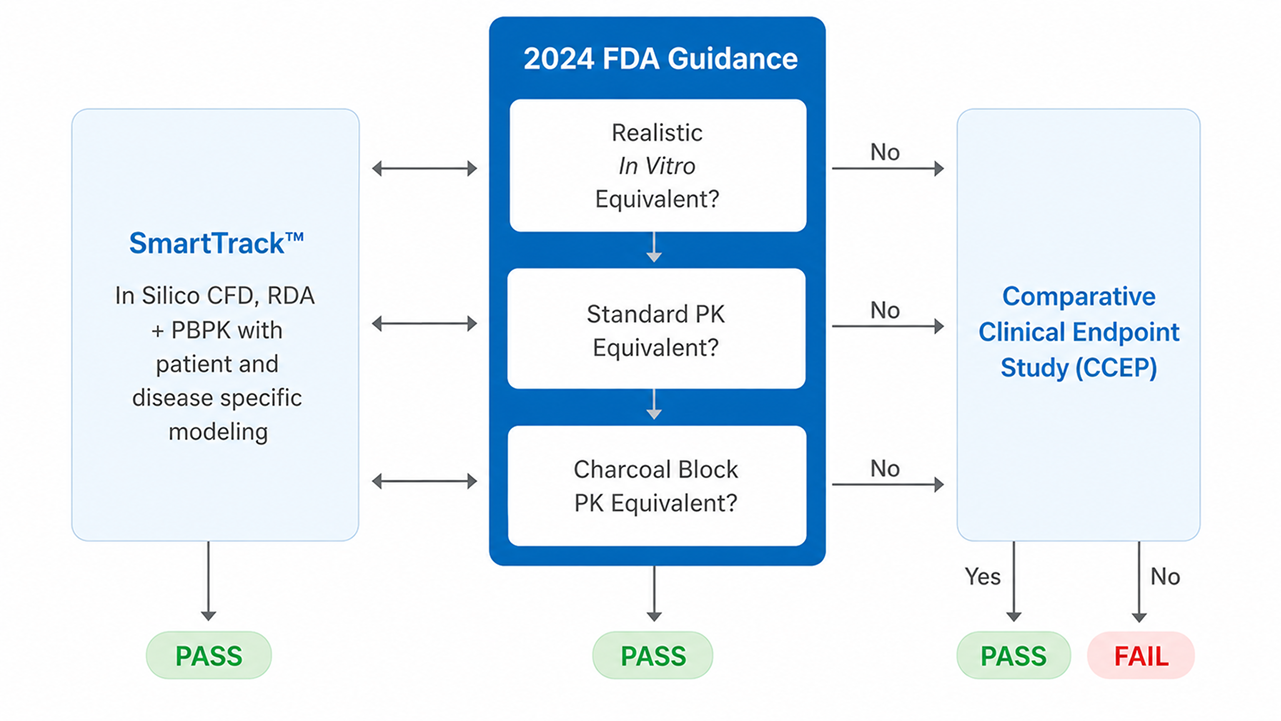
If any of the mandatory elements were to fail, SmartTrack™ can be applied to demonstrate whether or not these differences would really impact the comparative performance in a clinical setting and potentially support a justification of bioequivalence without going back to CCEP studies. Without computational modelling approaches, there is no alternative mitigation if there are any failures in the in vitro or clinical segments. Doing this proactively, rather than reactively, will derisk and accelerate the inhaled drug product development program.
Nanopharm has engaged with the U.S. FDA through sponsored workshops, research grants and broader collaborations for over 10 years to help better understand and define alternatives approaches to CCEP studies s for inhaled drug products.
along with Nanopharm and FLUIDDA to build its knowledge around modelling for OINDPs. These studies include , including for a next generation propellant (NGP) pressurized metered dose inhaler (pMDI) study.
have been held with Pharma Sponsors to move towards alignment on alternative approaches to bioequivalence, including for lower Global Warming Potential (GWP) propellant driven pMDIs.